Author: Denis Avetisyan
Researchers have developed a new machine learning framework that optimizes electronic structure calculations for crystalline materials, promising more accurate and efficient simulations.
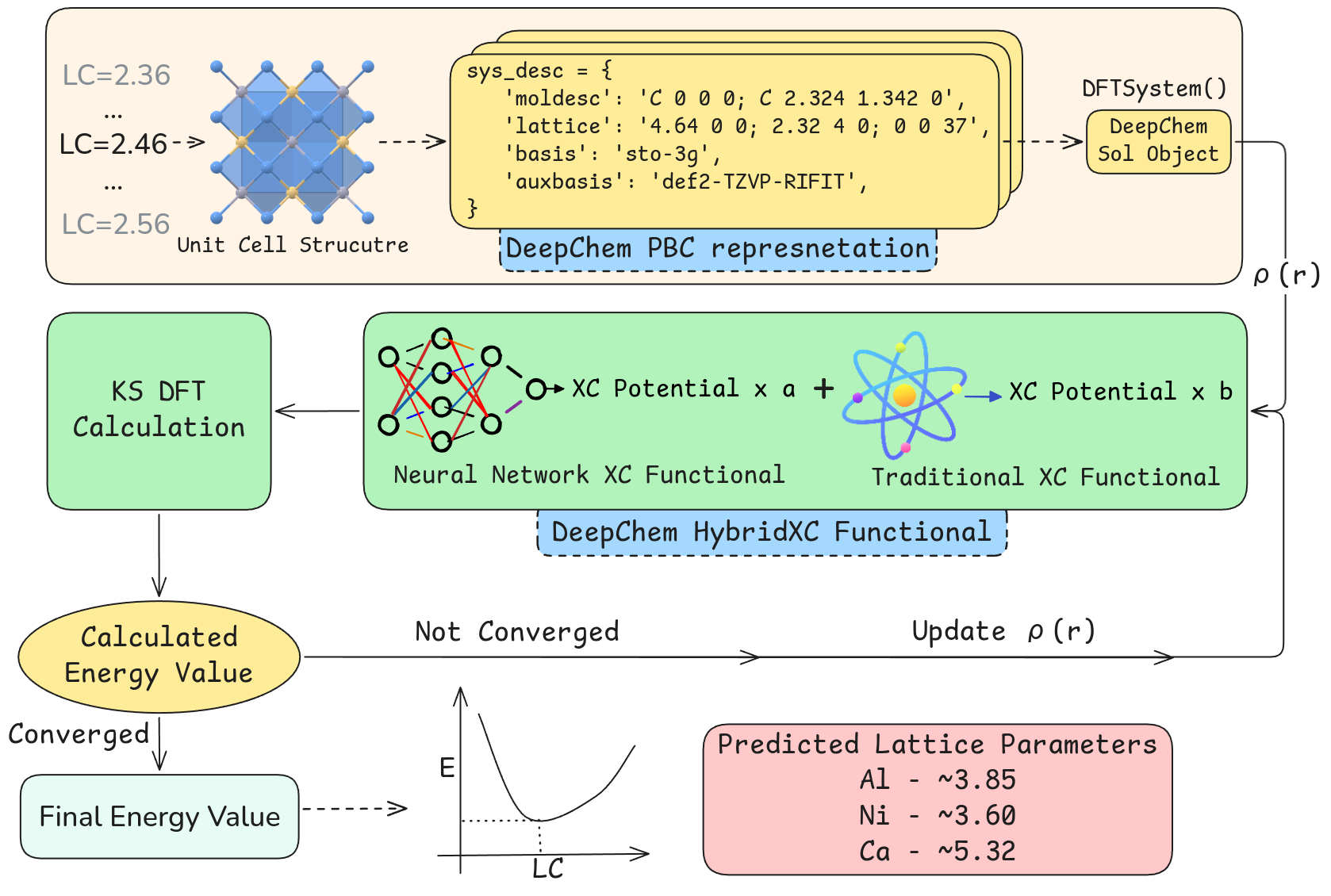
A fully differentiable approach enables the training of exchange-correlation functionals using neural networks within periodic boundary conditions.
Despite the widespread utility of Density Functional Theory (DFT) in materials science and chemistry, its computational expense remains a significant bottleneck for simulating large systems. This limitation motivates the development of more efficient exchange-correlation functionals, and this paper introduces ‘A fully differentiable framework for training proxy Exchange Correlation Functionals for periodic systems’ to address this challenge. By integrating machine learning models, specifically neural networks, into the DFT workflow and enabling end-to-end differentiability, the framework facilitates the training of improved functionals for periodic systems with relative errors as low as 5-10%. Will this approach pave the way for accurate and scalable electronic structure calculations, unlocking new possibilities in materials discovery and design?
The Computational Bottleneck: Why Materials Discovery Isn’t Scaling
Materials science increasingly depends on computational modeling to predict material properties and accelerate discovery, with Density Functional Theory (DFT) serving as the cornerstone for these simulations. However, DFT’s accuracy stems from a rigorous mathematical treatment of quantum mechanics, demanding significant computational resources – often scaling exponentially with system size. This presents a fundamental bottleneck, limiting the number of atoms and the duration of simulations that are practically feasible. Consequently, researchers are often restricted to modeling relatively small systems or simplified scenarios, hindering the ability to accurately predict the behavior of complex, real-world materials. The high computational cost effectively confines materials discovery to a limited ‘search space’, slowing the pace of innovation and preventing the exploration of potentially groundbreaking materials.
The fundamental challenge in accurately simulating materials at the atomic level lies in the intricate behavior of electrons, specifically the phenomenon of electron correlation. Describing how electrons interact with each other-beyond simple average-field approximations-is computationally demanding. Traditional Density Functional Theory (DFT) methods, while widely used, often struggle to capture these complex correlations with sufficient accuracy, leading to discrepancies between simulation results and experimental observations. To compensate for these limitations, researchers frequently resort to more sophisticated-and significantly more expensive-computational approaches or empirical corrections. This need for increased precision directly translates into a substantial demand for computational resources, limiting the size of systems and timescales that can be realistically modeled, and ultimately hindering the efficient discovery of novel materials with tailored properties.
The identification of materials possessing specific, tailored properties-critical for advancements in fields ranging from energy storage to aerospace engineering-is fundamentally constrained by the sheer computational expense of accurately predicting material behavior. Exhaustive screening, where a vast chemical space of potential materials is evaluated, demands an impractical number of calculations using methods like Density Functional Theory (DFT). Even with high-performance computing, the time required to assess each material’s characteristics-its stability, electronic structure, and mechanical properties-quickly becomes prohibitive, effectively limiting innovation to a small fraction of theoretically possible compounds. This bottleneck prevents researchers from fully exploring the potential of materials discovery, hindering the rapid development of next-generation technologies and leaving a vast landscape of potentially groundbreaking materials unexplored.
The escalating demand for novel materials with tailored properties is increasingly met with a computational barrier, as traditional Density Functional Theory (DFT) calculations – while accurate – demand significant resources. Machine learning surrogate models are emerging as a powerful solution, offering a means to predict material properties with accuracy approaching DFT, but at a drastically reduced computational cost. These models are trained on a relatively small dataset of DFT calculations, learning the complex relationships between a material’s atomic structure and its resulting properties. Once trained, they can rapidly evaluate the properties of countless candidate materials, effectively mapping out vast chemical spaces that would be inaccessible with conventional methods. This accelerated screening process promises to significantly shorten the materials discovery timeline, enabling researchers to identify promising candidates for experimental validation with unprecedented efficiency and paving the way for the design of materials with optimized functionalities.
Accelerating Electronic Structure: The Promise of Deep Learning
Deep learning frameworks, notably DeepChem and PyTorch, facilitate the construction of machine learning surrogate models for computationally expensive electronic structure calculations. These surrogate models, typically neural networks, are trained on datasets generated by established methods like Density Functional Theory (DFT). The resulting models approximate the input-output mapping of the full electronic structure calculation, enabling significantly faster prediction of material properties. DeepChem provides tools specifically tailored for representing and manipulating molecular and solid-state data, while PyTorch offers flexible tensor computation and automatic differentiation crucial for training complex neural network architectures.
Neural Exchange Correlation Functionals (NXCFs) represent a class of machine learning models designed to approximate the exchange-correlation functional within Density Functional Theory (DFT). These functionals, traditionally derived from complex many-body physics, are computationally expensive to evaluate. NXCFs, typically implemented as deep neural networks, are trained on datasets generated from high-fidelity DFT calculations, learning the mapping between atomic structure-defined by atomic coordinates and species-and resulting electronic properties such as energies, forces, and dipole moments. The neural network learns to predict the exchange-correlation energy E_{xc}[n] given the electron density n, effectively replacing the traditional, often approximated, functional form with a data-driven model. This allows for significantly faster prediction of electronic structure, with accuracy dependent on the size and quality of the training data and the network architecture.
Effective representation of solid-state systems is critical for the performance of deep learning models applied to electronic structure. The Sol Class within the DeepChem framework addresses this need by providing native support for representing solids, including features for handling periodic boundary conditions and crystallographic symmetry. This allows for the direct input of solid-state structures, such as those described by space groups, into neural networks. The Sol Class facilitates the calculation and inclusion of relevant descriptors – atomic coordinates, lattice vectors, and species – enabling the model to learn relationships between structure and properties without requiring computationally expensive pre-processing or feature engineering specific to solid-state materials. This native framework significantly streamlines the development and training process for machine learning models targeting solid-state systems.
Density Functional Theory (DFT) based on the Kohn-Sham equations constitutes the primary method for generating the training datasets used in machine learning accelerated electronic structure calculations. These calculations, performed with established codes, yield high-fidelity data representing the relationship between atomic structure and resulting electronic properties, such as energies and forces. The accuracy of the surrogate models-typically neural networks-is directly dependent on the quality and quantity of this DFT-derived data; larger and more diverse datasets generally lead to improved model performance and generalization capabilities. The computational expense of DFT necessitates careful consideration of dataset size and the selection of representative structures for training, often employing techniques like active learning to maximize data efficiency.
Bridging Accuracy and Efficiency: Hybrid Functionals in Action
The HybridXC class of functionals represents a machine learning approach to density functional theory (DFT) that integrates established Generalized Gradient Approximation (GGA) functionals with Multi Layer Perceptrons (MLPs). GGAs, while computationally efficient, can exhibit limitations in accurately describing complex electronic interactions. HybridXC addresses this by employing MLPs to learn corrections to the GGA functional, effectively enhancing its predictive power. The MLP component provides a flexible, data-driven means of capturing non-local correlation effects not inherently accounted for in standard GGA approximations, while retaining the computational benefits associated with the underlying GGA framework. This allows for improvements in accuracy without the substantial computational cost typically associated with more advanced methods like Coupled Cluster theory.
HybridXC functionals enhance the accuracy of Generalized Gradient Approximation (GGA) density functionals by incorporating machine learning corrections. Rather than replacing the GGA functional entirely, the model learns to predict and apply corrections to the GGA output, thereby improving the overall energy and structural predictions. This approach minimizes computational overhead because the majority of the calculation still relies on the efficient GGA framework; the machine learning component introduces a comparatively small additional cost. Consequently, HybridXC functionals offer a favorable trade-off between accuracy and computational efficiency, making them suitable for larger systems where high-level, but computationally expensive, methods are impractical.
The generation of training data for HybridXC models relies on Kohn-Sham Density Functional Theory (DFT) calculations. A minimal, yet effective, approach utilizes the PBE functional, a commonly employed Generalized Gradient Approximation (GGA), in conjunction with the STO-3G basis set. This combination provides a computationally efficient method for generating a substantial dataset of electronic structure properties. While larger basis sets and more sophisticated functionals could be used, the STO-3G/PBE pairing offers a favorable balance between accuracy and computational cost, enabling the creation of a dataset sufficiently large for training the Multi Layer Perceptron component of the HybridXC functional.
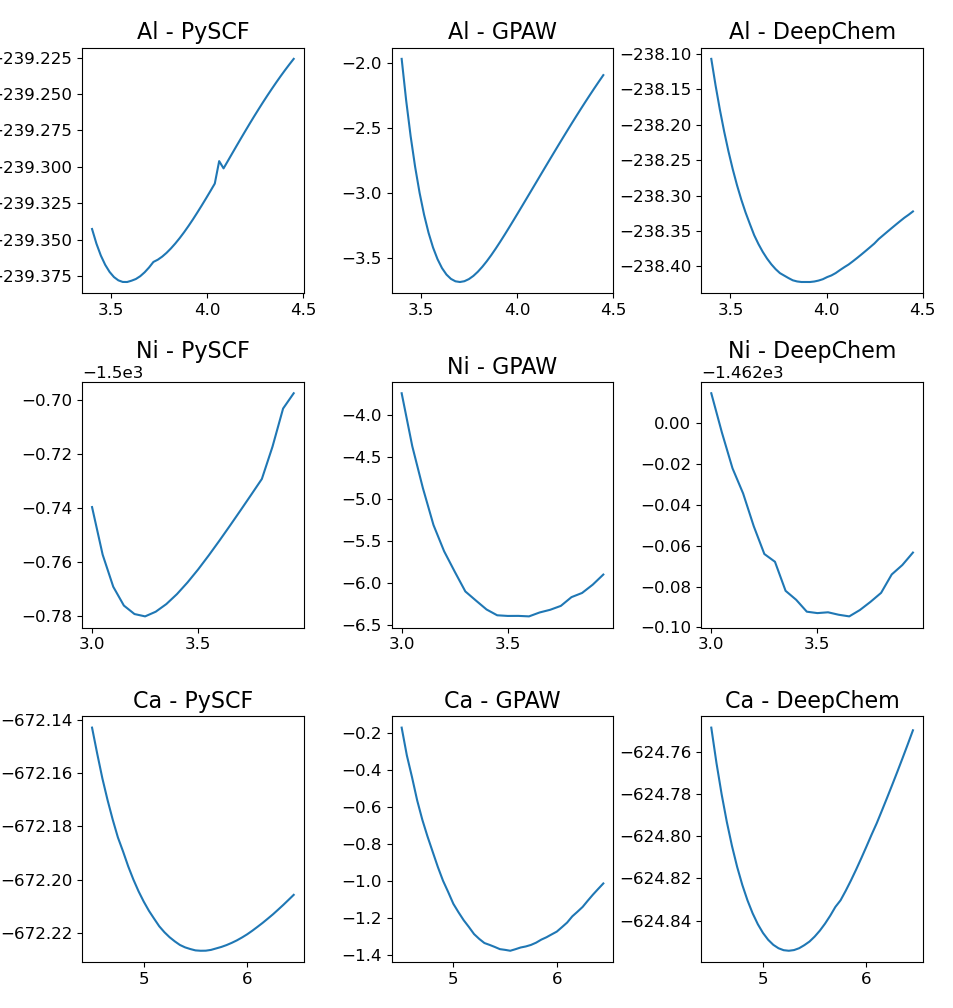
Model validation utilizes lattice constant optimization performed on face-centered cubic systems to assess predictive accuracy. Comparisons to reference calculations, generated with established density functional theory packages such as GPAW and PySCF, indicate a relative error of approximately 5-10% in predicted lattice constants. This error range was determined through systematic comparison of optimized lattice parameters calculated by the HybridXC model against those computed using established DFT methods and basis sets, providing a quantitative measure of the model’s performance on this specific structural property.
Scaling Materials Discovery: A Future Forged in Data and Efficiency
The continued development of accurate materials modeling relies heavily on the availability of robust, experimentally-derived datasets, and the Materials Project offers an exceptionally valuable resource in this regard. This open-access database contains a wealth of computed and experimental data on known materials, providing a crucial benchmark for validating the predictive power of new models and refining their parameters. By training algorithms on this extensive collection of real-world data, researchers can significantly improve the reliability and accuracy of materials predictions, moving beyond theoretical approximations toward a more data-driven approach. Furthermore, the Materials Project’s continuously expanding dataset allows for ongoing assessment and improvement of model performance, fostering a cycle of iterative refinement that accelerates the discovery of novel materials with desired properties.
The current model, while effective, faces limitations when applied to increasingly complex materials systems. To address this, researchers are actively transitioning from Gaussian basis sets – which describe atomic orbitals – to Plane Wave Basis sets. This shift isn’t merely a technical adjustment; it fundamentally alters the computational landscape. Plane Wave Basis sets, while requiring more computational resources initially, scale more favorably with system size. This means that as the number of atoms in the modeled material increases, the computational cost grows at a slower rate compared to Gaussian basis sets. Consequently, the adoption of Plane Wave Basis sets promises to unlock the ability to accurately simulate vastly larger and more intricate materials, including those with complex crystal structures and compositions, ultimately accelerating the discovery of novel materials with tailored properties.
The developed framework offers a pathway to dramatically expedite materials discovery through the efficient exploration of expansive chemical spaces. Traditionally, identifying novel materials with desired properties involved painstaking trial-and-error, often limited by computational expense. This new approach, however, facilitates high-throughput screening – the automated prediction of material properties for countless candidate compounds. By swiftly narrowing the field of possibilities, researchers can focus experimental efforts on the most promising candidates, significantly reducing both time and resource investment. The ability to virtually ‘test’ materials before synthesis promises to unlock innovations across diverse fields, from energy storage and catalysis to advanced electronics and structural materials, fostering a new era of accelerated materials innovation.
A significant reduction in the computational expense of materials modeling promises to revolutionize the field of materials design. Currently, predicting the properties of novel materials often requires extensive and time-consuming simulations, limiting the scope of exploration. Lowering these computational barriers enables researchers to virtually screen an unprecedented number of material candidates, accelerating the identification of compounds with desired characteristics – from high-strength alloys for aerospace applications to efficient semiconductors for renewable energy technologies and novel catalysts for chemical processes. This capability extends beyond simply finding better materials; it facilitates the inverse design process, where materials are specifically engineered to achieve targeted functionalities, opening doors to customized solutions for a diverse range of technological challenges and ultimately fostering innovation across multiple scientific disciplines.
The pursuit of accurate electronic structure calculations, as detailed in this work, reveals a fundamental truth about modeling: it is not about achieving perfect rationality, but about managing inherent imperfections. The framework presented attempts to refine existing exchange-correlation functionals, acknowledging their limitations and seeking incremental improvements through machine learning. This resonates with the ancient wisdom of Epicurus, who observed, “It is not the pursuit of pleasure itself that is wrong, but the pursuit of it at the expense of tranquility.” Similarly, this research doesn’t aim for absolute perfection in calculations, but for a more stable, efficient, and ultimately, tranquil approach to understanding complex crystalline materials. The model builders recognize that even the most sophisticated algorithms are built upon approximations, mirroring the human tendency to prioritize workable solutions over unattainable ideals.
What Lies Ahead?
The pursuit of more accurate exchange-correlation functionals, even within a machine learning framework, feels less like a technical challenge and more like a refinement of existing biases. The framework presented here, while elegant, doesn’t eliminate the need for initial guesses – it simply offers a more efficient way to nudge those guesses toward agreement with observed data. Even with perfect information, people choose what confirms their belief; models are no different. The true limitation isn’t computational cost, but the inherent difficulty in escaping the pre-conceptions embedded within the training data itself.
Future work will undoubtedly focus on expanding the scope of these differentiable models to include dynamical effects and excited-state properties. However, a more pressing question is whether increasing complexity will truly yield insight, or merely provide more convincing illusions. Most decisions aim to avoid regret, not maximize gain, and the same principle applies to model construction. Researchers will naturally gravitate toward improvements that minimize immediate error, potentially overlooking more fundamental, but less readily quantifiable, shortcomings.
Ultimately, the success of this approach, and others like it, will depend not on achieving perfect predictive power, but on providing a framework for systematically understanding where these models fail. The goal isn’t to build a perfect simulation of reality, but a more honest reflection of the assumptions – and the inherent limitations – that underpin it.
Original article: https://arxiv.org/pdf/2602.15923.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- 20 Movies Where the Black Villain Was Secretly the Most Popular Character
- Celebs Who Narrowly Escaped The 9/11 Attacks
- Top 20 Dinosaur Movies, Ranked
- 25 “Woke” Films That Used Black Trauma to Humanize White Leads
- Transformers Under the Microscope: What Graph Neural Networks Reveal
- The 10 Most Underrated Jim Carrey Movies, Ranked (From Least to Most Underrated)
- 22 Films Where the White Protagonist Is Canonically the Sidekick to a Black Lead
- Trading on Thin Air: AI Agents Conquer Crypto Volatility
- The Best Directors of 2025
- Gold Rate Forecast
2026-02-19 20:41