Author: Denis Avetisyan
Researchers have harnessed the power of graph neural networks to rapidly predict the infrared spectra of polycyclic aromatic hydrocarbons, the molecules that permeate interstellar space.
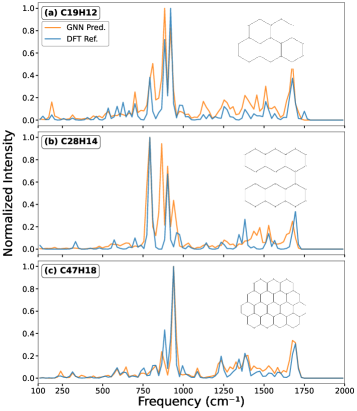
This work demonstrates accurate and efficient spectral prediction using the Attentive Fingerprint architecture, offering a significant advantage over traditional quantum chemical calculations for complex molecules.
Despite the crucial role of polycyclic aromatic hydrocarbons (PAHs) in driving the aromatic infrared bands observed throughout the interstellar medium, computationally intensive quantum chemical calculations have historically limited our ability to interpret these spectra given the vast structural diversity of PAH molecules. This work, ‘Graph Neural Network Prediction of Infrared Spectra of Interstellar Polycyclic Aromatic Hydrocarbons’, introduces a graph neural network framework-specifically an Attentive Fingerprint model-capable of predicting PAH infrared spectra up to 10,000 times faster than traditional methods. Our results demonstrate that this approach yields accurate predictions for PAHs containing 20-40 carbon atoms, achieved through training with the Jensen-Shannon divergence as a loss function. Could this efficient spectral prediction framework unlock new avenues for analyzing complex astronomical spectra and furthering our understanding of interstellar chemistry?
The Unseen Echoes of the Cosmos
Across the cosmos, astronomical spectra are punctuated by a series of mysterious emissions in the infrared region – collectively known as Unidentified Infrared Emission (UIE). These features aren’t attributable to known atomic or molecular species, suggesting the presence of complex molecules previously undetected in interstellar space. The pervasiveness of UIE – observed in planetary nebulae, reflection nebulae, and even distant galaxies – indicates these molecules are not rare exotics, but rather a significant, though elusive, component of the interstellar medium. Their broad spectral signatures hint at structures far more intricate than simple molecules, prompting investigations into increasingly complex carbon-based compounds as potential sources. Understanding the origin of UIE is therefore crucial not only for deciphering the chemical composition of space, but also for a more complete understanding of the lifecycle of stars and the formation of planetary systems.
The pervasive presence of unidentified infrared emission (UIE) features in astronomical observations strongly suggests the existence of complex molecules throughout interstellar space, and polycyclic aromatic hydrocarbons (PAHs) currently represent the most promising explanation. However, definitively confirming PAHs as the source of these signals demands highly accurate spectral modeling – a process that involves predicting the infrared light emitted by these molecules under various astrophysical conditions. This isn’t simply a matter of identifying the presence of PAHs; the precise shape and intensity of the UIE features are sensitive to a molecule’s size, structure, and chemical environment. Therefore, detailed simulations are essential to compare predicted spectra with observed data, allowing astronomers to constrain the properties of PAHs in space and rule out alternative explanations for the mysterious infrared glow.
Calculating the infrared spectra of polycyclic aromatic hydrocarbons (PAHs) – molecules proposed to explain a significant portion of unidentified infrared emission in space – presents a substantial computational challenge. Traditional methods for these calculations scale with a complexity of N_C^{4.18}, where N_C represents the number of carbon atoms in the PAH molecule. This steep scaling relationship means that even modest increases in molecular size dramatically increase the computational resources required, quickly rendering analysis of larger, more complex PAHs – which are likely present in astrophysical environments – virtually impossible. Consequently, astronomers are limited in their ability to accurately model and interpret observed spectra, hindering efforts to definitively confirm the role of PAHs in explaining the ubiquitous unidentified infrared emission and fully characterize the molecular composition of interstellar space.
From Molecular Fingerprints to Computational Shadows
Molecular fingerprints represent a method of discretizing the structural information of Polycyclic Aromatic Hydrocarbons (PAHs) into a fixed-length bit string. These fingerprints encode the presence or absence of specific substructures, functional groups, or topological features within the PAH molecule. Early machine learning models leveraged these fingerprints as input features, effectively treating PAH structure as a series of binary characteristics. This approach simplified the complex structural data, enabling computational analysis despite the inherent limitations of representing continuous molecular properties with discrete values. The resulting fingerprint vectors served as a computationally efficient means of quantifying structural similarity and diversity within the NASA Ames PAH Database and facilitated the training of predictive models.
Random Forest Models and Feedforward Neural Networks were successfully applied to the prediction of Polycyclic Aromatic Hydrocarbon (PAH) spectra based on their molecular structures, establishing the potential of machine learning in this domain. These models utilized algorithms to identify correlations between structural features-such as the number of fused rings and the presence of specific functional groups-and resulting spectral characteristics like peak positions and intensities. Early implementations achieved predictive accuracy sufficient to demonstrate the feasibility of learning these structure-spectrum relationships, although limitations existed in capturing the full complexity of spectral data generated by larger, more intricate PAH molecules.
Early machine learning models, while demonstrating predictive capability for Polycyclic Aromatic Hydrocarbon (PAH) spectra, exhibited limitations in accurately representing the complexities of larger, more structurally diverse PAHs. These models often failed to fully resolve fine spectral details, particularly in the infrared region, due to an inability to account for subtle interactions between vibrational modes and the influence of extended π systems. The simplified representations of PAH structure used as input, such as molecular fingerprints, lacked the granularity necessary to capture the nuances arising from variations in PAH size, shape, and the presence of heteroatoms, leading to reduced predictive power for complex molecules and an underestimation of spectral feature intensities.
The NASA Ames PAH Database is a computationally derived collection of predicted spectra for over 180 polycyclic aromatic hydrocarbon (PAH) molecules and their ions. These data were generated using density functional theory (DFT) calculations, specifically the B3LYP functional with the 6-311+G(d,p) basis set, to simulate the electronic transitions responsible for the observed ultraviolet-visible spectra. The database provides accurate theoretical spectra, including transition energies and oscillator strengths, which serve as essential training and validation data for machine learning models attempting to correlate PAH structure with spectral features. Its publicly available format and comprehensive scope have made it a foundational resource for the astrochemical community and a key component in the development of predictive PAH spectral analysis tools.
Graph Neural Networks: A New Spectral Paradigm
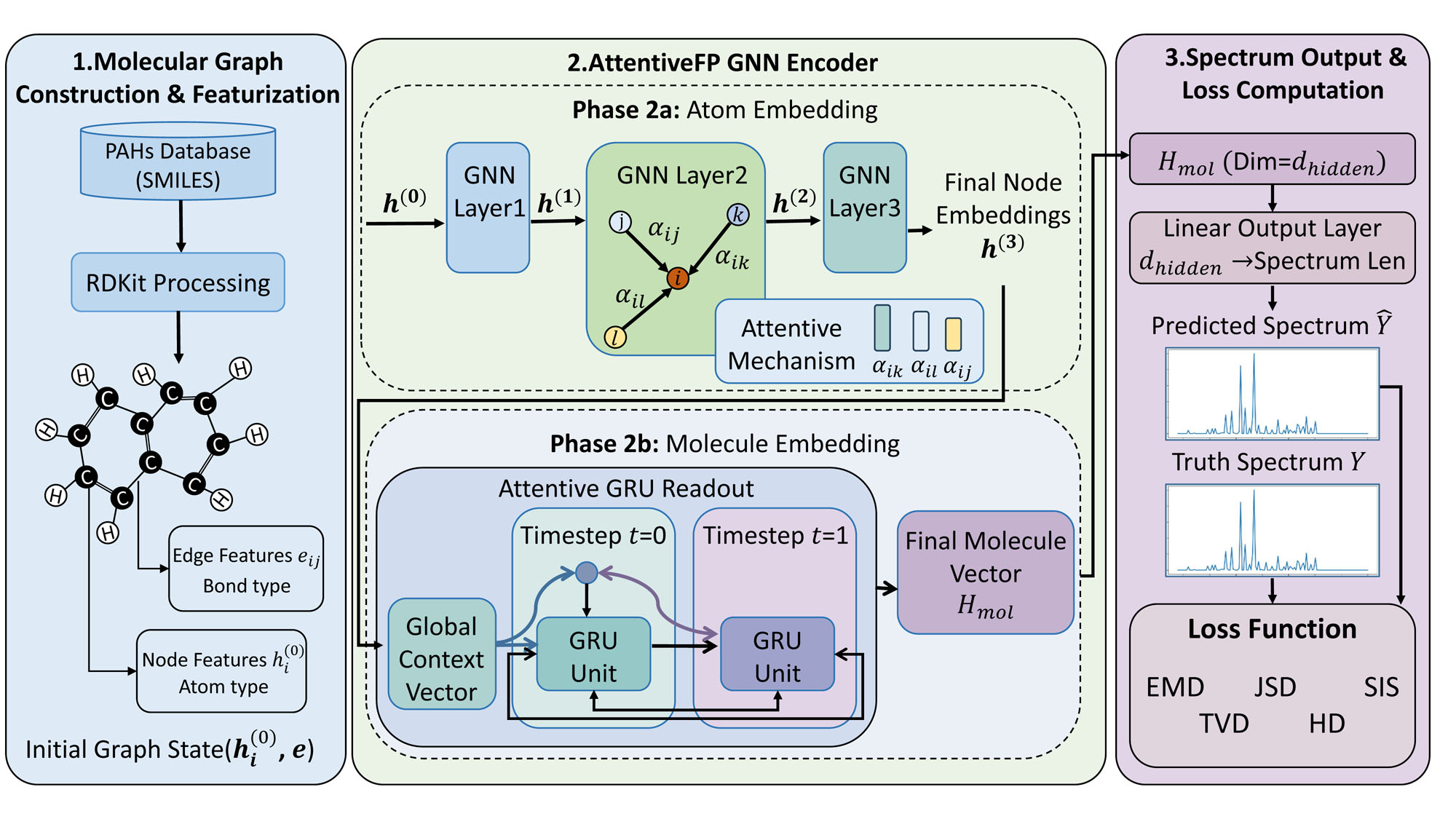
Graph Neural Networks (GNNs) represent molecular structures as graphs, where atoms are nodes and chemical bonds are edges, allowing the model to directly incorporate structural information into its calculations. This approach contrasts with traditional methods that rely on representations like SMILES strings or fingerprints which can lose explicit connectivity data. By encoding this structural connectivity, GNNs can more effectively predict molecular properties – specifically spectral characteristics – as these properties are directly influenced by the arrangement of atoms and bonds within the molecule. The graph representation enables the application of graph convolution and message passing algorithms, allowing information to propagate between bonded atoms and facilitating the learning of complex relationships between molecular structure and spectral output.
Graph Convolutional Networks (GCNs), Message Passing Neural Networks (MPNNs), and Graph Attention Networks (GATs) all utilize graph representations of molecular structures to extract and learn complex spectral features. GCNs apply convolution operations directly on the graph structure, aggregating information from neighboring nodes to learn node embeddings. MPNNs generalize this approach by defining a message-passing framework with learnable message functions and update functions, allowing for greater flexibility in feature aggregation. GATs introduce attention mechanisms, weighting the contributions of neighboring nodes based on their relevance, thereby enabling the model to focus on the most important structural features for spectral prediction. These architectures effectively transform the graph structure into learned feature vectors suitable for downstream tasks like property prediction or spectra reconstruction.
Simplified Molecular Input Line Entry System (SMILES) strings are a linear notation for representing molecular structures and are crucial for interfacing molecules with Graph Neural Network (GNN) architectures. These strings, consisting of characters denoting atoms and bonds, provide a standardized, text-based format that can be readily parsed and converted into a graph representation suitable for GNNs. This process involves mapping atoms to nodes and bonds to edges, effectively translating the 1D SMILES string into the 2D or 3D graph structure required by the GNN for spectral property prediction. The use of SMILES streamlines data input, allowing for automated processing of large molecular datasets without the need for manual graph construction.
Attentive Fingerprint is a Graph Neural Network (GNN) architecture designed to enhance spectral prediction accuracy through the implementation of attention mechanisms. These mechanisms allow the network to differentially weight the importance of various atoms and bonds within a molecular graph during the message-passing process. By focusing on the most relevant structural features, Attentive Fingerprint can learn more refined representations of molecular properties. The attention weights are learned during training, enabling the model to automatically identify key substructures that contribute significantly to the target spectral characteristics. This approach improves performance compared to GNNs with uniform weighting schemes and facilitates the interpretation of model predictions by highlighting influential molecular fragments.
Quantifying the Shadows: Spectral Similarity Metrics
Quantitative comparison of predicted and observed spectra necessitates the application of robust distance metrics. These metrics serve to calculate the dissimilarity between two spectral datasets, enabling objective assessment of prediction accuracy. The selection of an appropriate metric is crucial, as different metrics emphasize varying aspects of spectral difference; for example, some prioritize differences in peak intensity, while others focus on shifts in peak position or overall spectral shape. Commonly employed metrics include those based on statistical divergence and information theory, which provide a quantifiable measure of the difference between the probability distributions represented by the spectra. The sensitivity and reliability of these metrics directly influence the validity of spectral predictions and subsequent data analysis.
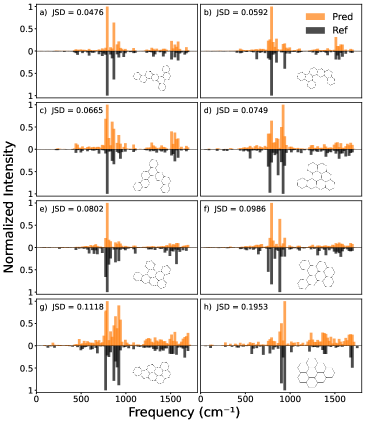
Several spectral distance metrics are employed to quantify the similarity between predicted and observed spectra, each offering a distinct approach to comparison. Earth Mover’s Distance (EMD) calculates the minimum “work” required to transform one spectrum into another, effectively measuring the cost of spectral feature displacement. Jensen-Shannon Divergence (JSD) provides a smoothed and symmetrized version of the Kullback-Leibler divergence, offering a probabilistic measure of dissimilarity. Hellinger Distance assesses the overlap between the square roots of spectral distributions, providing a bounded and stable metric. Total Variation Distance calculates half the sum of the absolute differences between spectral values, representing the maximum possible difference between the two spectra. Finally, Spectrum Information Similarity (SIS) focuses on the shared information content between spectra, offering a measure of their correlation. These metrics each provide a unique perspective on spectral dissimilarity and are chosen based on the specific characteristics of the data and the desired sensitivity to spectral features.
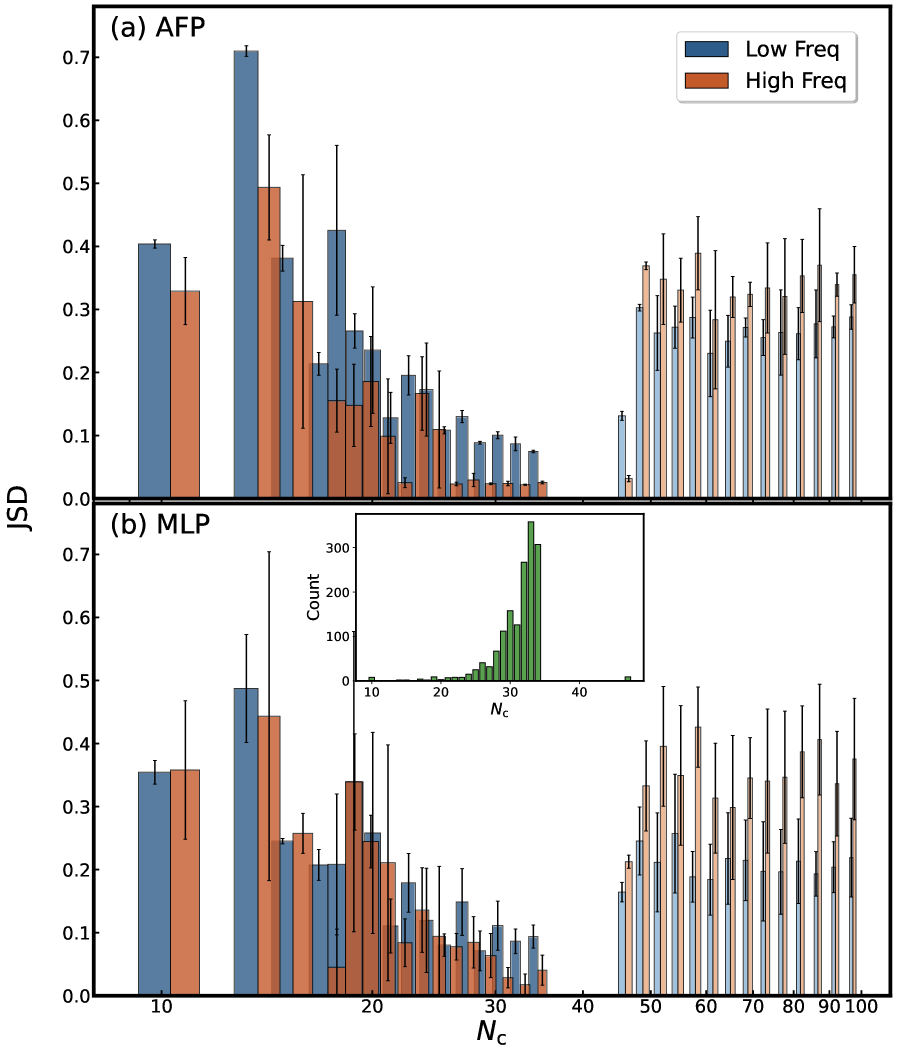
Attentive Fingerprint employs spectral distance metrics – including Earth Mover’s Distance, Jensen-Shannon Divergence, Hellinger Distance, Total Variation Distance, and Spectrum Information Similarity – directly within its network architecture as loss functions. This implementation moves beyond traditional mean squared error approaches by quantifying the dissimilarity between predicted and observed spectra using these specialized metrics. By minimizing these distance metrics during training, the network is optimized to generate predicted spectra that more closely resemble the observed spectra, improving the accuracy of spectral reproduction and, consequently, the ability to identify and characterize polycyclic aromatic hydrocarbons (PAHs).
The accuracy of polycyclic aromatic hydrocarbon (PAH) identification and characterization in astronomical spectra is directly contingent upon the selected spectral distance metric. These metrics quantify the dissimilarity between observed and predicted spectra, and their sensitivity impacts the ability to resolve subtle spectral features indicative of specific PAH molecules and their properties-such as ionization state and size. Insufficient metric sensitivity can lead to misidentification or an inability to distinguish between different PAHs, while overly sensitive metrics may be affected by noise or other spectral artifacts. Consequently, the choice of metric-including Earth Mover’s Distance, Jensen-Shannon Divergence, and others-is a crucial factor in extracting reliable PAH information from astronomical datasets and building accurate spectral models.
Unveiling the Interstellar Tapestry
A notable advancement in astrochemistry stems from the confluence of Graph Neural Networks (GNNs) and refined spectral metrics, dramatically enhancing the modeling of Polycyclic Aromatic Hydrocarbon (PAH) spectra. Traditionally, simulating these complex spectra relied heavily on Density Functional Theory (DFT) calculations – a computationally expensive process that scales unfavorably with molecular size. This new approach circumvents those limitations by leveraging the ability of GNNs to learn directly from the underlying molecular structure, offering a substantial speedup and reduced computational complexity. The resulting models can rapidly and accurately predict PAH spectra, enabling researchers to more effectively analyze astronomical observations and unlock crucial details regarding the composition and physical conditions of interstellar and circumstellar environments, ultimately providing a deeper understanding of the interstellar medium’s evolution.
The ability to accurately model polycyclic aromatic hydrocarbon (PAH) spectra is paramount to deciphering the enigmatic Unidentified Infrared Emission (UIE) features pervasive throughout interstellar and circumstellar spaces. These UIE bands, observed across a wide range of astronomical sources, represent a significant fraction of the total infrared luminosity of galaxies, yet their precise origins have long remained a mystery. Detailed spectral modeling allows researchers to move beyond simply detecting PAHs – ubiquitous molecules formed in the harsh conditions of space – and instead determine their specific size, structure, and ionization state. This, in turn, provides crucial insights into the physical conditions – such as temperature, density, and radiation field strength – within star-forming regions, protoplanetary disks, and the broader interstellar medium, ultimately illuminating the processes of cosmic evolution and the potential for complex chemistry beyond our solar system.
Polycyclic aromatic hydrocarbons (PAHs) are pervasive throughout the interstellar medium, and their precise identification and quantification offer a powerful window into the composition and life cycle of this crucial cosmic component. These molecules, formed in the outflows of dying stars and in the harsh environments of supernova remnants, contribute significantly to the overall abundance of carbon in galaxies. By meticulously cataloging the variety and prevalence of PAHs – from small molecules containing a few carbon atoms to larger, more complex structures – scientists can trace the pathways of carbon processing in space. This detailed analysis reveals how elements are cycled between stars and interstellar gas, providing critical data for understanding galactic chemical evolution and the conditions necessary for planet formation. Consequently, a refined ability to characterize PAHs isn’t simply about identifying molecules; it’s about deciphering the very building blocks and evolutionary history of galaxies themselves.
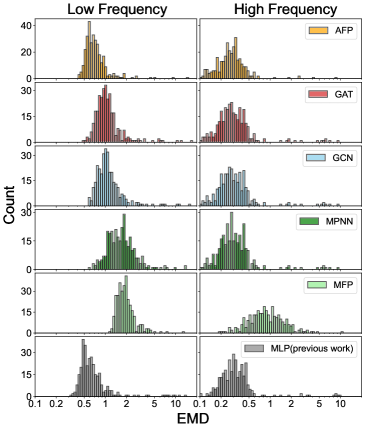
A novel computational approach, the Attentive Fingerprint model, dramatically accelerates the analysis of polycyclic aromatic hydrocarbons (PAHs) – crucial components of the interstellar medium. This model achieves processing speedups of two to five orders of magnitude when compared to traditional Density Functional Theory (DFT) calculations, largely due to its significantly improved computational scaling – N_C^{0.21} versus DFT’s N_C^{4.18}, where N_C represents the number of carbon atoms. Rigorous validation, employing the Earth Mover’s Distance as a metric for accuracy, demonstrates that the Attentive Fingerprint model consistently surpasses the performance of other graph neural network baselines, particularly when analyzing intermediate-sized PAHs containing between 21 and 34 carbon atoms; this enhanced efficiency and accuracy promise to unlock new possibilities in interpreting complex spectral data from interstellar and circumstellar environments.

The pursuit of spectral prediction for polycyclic aromatic hydrocarbons, as detailed in this work, highlights the inherent limitations of even the most sophisticated theoretical frameworks. The Attentive Fingerprint architecture, while offering a remarkable acceleration over quantum chemical calculations, still faces hurdles when extrapolating to larger molecular structures. This mirrors a fundamental truth: any model, no matter how elegantly constructed, is ultimately an approximation of reality. As Pyotr Kapitsa once observed, “It is better to be able to explain something than to know it.” This research embodies that sentiment; it prioritizes predictive capability, even while acknowledging the potential for theoretical boundaries, reminding one that black holes are the best teachers of humility; they show that not everything is controllable.
Where Do the Spectra Lead?
The efficient prediction of infrared spectra for polycyclic aromatic hydrocarbons, as demonstrated, offers a fleeting glimpse of order within a fundamentally chaotic system. Any model, however sophisticated, remains an approximation – a map, not the territory. The true spectra of these interstellar molecules, sculpted by radiation fields and stochastic collisions, likely harbor subtleties beyond the reach of even the most attentive graph neural network. The current limitations in extrapolating to exceedingly large PAH structures serve as a useful reminder: any hypothesis about molecular complexity is merely an attempt to hold infinity on a sheet of paper.
Future efforts will undoubtedly focus on expanding the training datasets and refining the network architectures. Yet, a more profound challenge lies in bridging the gap between computational prediction and astrophysical observation. The spectral signatures detected in space are rarely pristine; they are redshifted, broadened, and superimposed upon a noisy background. To truly decode the messages carried by these molecules requires not just accurate models, but also a rigorous understanding of the environments in which they reside.
Black holes teach patience and humility; they accept neither haste nor noise. This work, while a technical advance, should be viewed not as a destination, but as a step toward a more nuanced appreciation of the universe’s inherent ambiguity. The spectra beckon, but the full story will likely remain forever beyond complete comprehension.
Original article: https://arxiv.org/pdf/2602.12560.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- 2025 Crypto Wallets: Secure, Smart, and Surprisingly Simple!
- Gold Rate Forecast
- Here Are the Best TV Shows to Stream this Weekend on Paramount+, Including ‘48 Hours’
- Top 15 Celebrities in Music Videos
- Top 20 Extremely Short Anime Series
- Where to Change Hair Color in Where Winds Meet
- 20 Films Where the Opening Credits Play Over a Single Continuous Shot
- Top gainers and losers
- 50 Serial Killer Movies That Will Keep You Up All Night
- 20 Must-See European Movies That Will Leave You Breathless
2026-02-16 15:18