Author: Denis Avetisyan
A new dynamic bootstrapping method improves the accuracy and efficiency of training machine learning interatomic potentials by actively filtering out noisy data during the learning process.
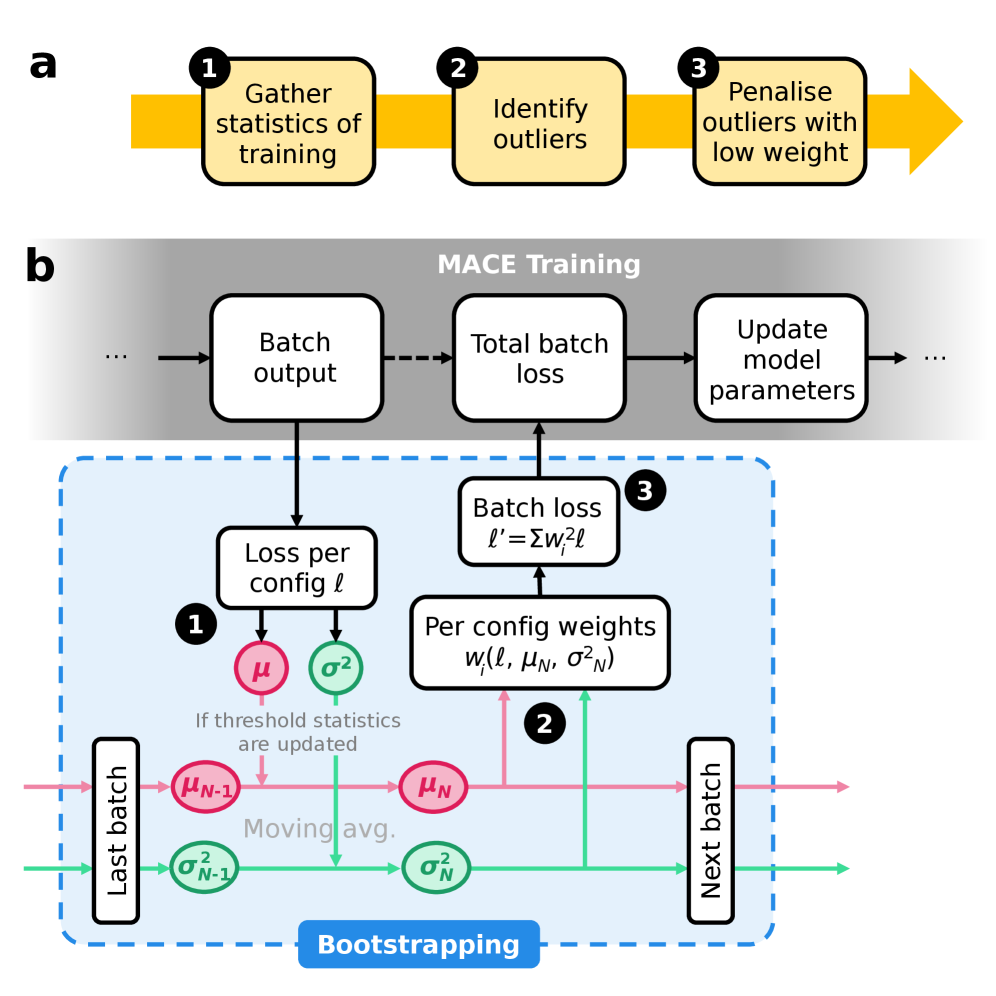
This review details an on-the-fly outlier detection technique for building robust foundation models used in molecular dynamics and active learning.
The accuracy of machine learning models is often undermined by imperfections in the training data itself. Addressing this challenge, our work, ‘Cutting Through the Noise: On-the-fly Outlier Detection for Robust Training of Machine Learning Interatomic Potentials’, introduces an automated scheme to dynamically down-weight noisy samples during the training of machine learning interatomic potentials. This on-the-fly outlier detection, leveraging an exponential moving average of the loss distribution, achieves performance comparable to iterative refinement with significantly reduced computational cost, as demonstrated by improvements in diffusion coefficients for liquid water and a threefold reduction in energy errors for a foundation model trained on the SPICE dataset. Could this approach unlock more robust and efficient training pipelines for machine learning models across diverse scientific domains and dataset scales?
The Challenge of Accurate Molecular Representation
High-fidelity electronic structure calculations, often achieved through methods like Ab Initio Quantum Chemistry, strive to solve the Schrödinger equation with minimal approximations, providing highly accurate descriptions of molecular systems. However, this accuracy comes at a steep price: computational cost that scales rapidly with system size-often factorially or even worse. Consequently, simulating even moderately sized molecules, let alone complex materials or biological assemblies, becomes prohibitively expensive, restricting the scope of investigations. The computational demands arise from the need to accurately represent the many-body interactions between electrons, requiring extensive calculations of integrals and wavefunctions. This limitation necessitates either the use of less accurate, faster methods or the development of innovative algorithms and hardware to tackle these computationally intensive problems, ultimately hindering the ability to model and predict the behavior of increasingly complex chemical and physical systems.
While offering a significant computational advantage over methods like Ab Initio quantum chemistry, Density Functional Theory (DFT) isn’t without its limitations. The accuracy of DFT hinges on the chosen exchange-correlation functional, an approximation of the complex many-body interactions governing electron behavior. Different functionals perform variably across different systems, and the lack of a universally accurate functional introduces potential for systematic errors. Furthermore, achieving convergence – obtaining a stable solution – can be problematic, particularly for systems exhibiting strong correlation or complex electronic structures. This sensitivity to both functional choice and convergence criteria directly impacts the reliability of results obtained from DFT calculations, demanding careful validation and consideration of potential uncertainties when interpreting predicted molecular properties and behaviors.
Stochastic methods, prominently including Variational Monte Carlo (VMC) and Diffusion Monte Carlo (DMC), introduce an unavoidable element of statistical noise into calculations of molecular properties. These techniques rely on random sampling to solve the many-body Schrödinger equation, meaning results aren’t deterministic but rather represent averages over numerous simulations. This inherent noise manifests as uncertainty in the computed reference data – even with extensive sampling, a precise solution remains elusive. The magnitude of this uncertainty depends on factors like the system’s complexity, the number of samples used, and the specific estimator employed. While sophisticated variance reduction techniques can mitigate the issue, the stochastic nature of these methods necessitates careful consideration of error bars and statistical significance when interpreting results and comparing them to experimental data or other computational approaches; a seemingly insignificant difference may simply fall within the bounds of statistical fluctuation.
Machine Learning Interatomic Potentials: A Pathway to Computational Efficiency
Machine Learning Interatomic Potentials (MLIPs) represent a computational method for predicting the potential energy of atomic systems, enabling accelerated molecular dynamics simulations. Traditional molecular dynamics relies on empirically derived potential functions, which often lack accuracy or transferability. MLIPs address these limitations by learning potential energy surfaces directly from data generated by accurate, first-principles calculations such as Density Functional Theory (DFT) or Coupled Cluster methods. These ab initio calculations, while highly accurate, are computationally expensive, limiting the timescale and system size of achievable simulations. MLIPs circumvent this bottleneck by training a machine learning model – typically a neural network – to approximate the potential energy surface, allowing for rapid and accurate predictions of atomic forces and energies at a fraction of the computational cost. The trained MLIP can then be used to perform long-timescale molecular dynamics simulations, effectively bridging the gap between accuracy and computational feasibility.
The performance of Machine Learning Interatomic Potentials (MLIPs) is directly correlated to the quality of the training data used in their development. While MLIPs aim to approximate potential energy surfaces from computationally expensive Ab Initio quantum chemistry calculations, these underlying calculations are not without error. Noise in the training data can originate from several sources including numerical tolerances within the Ab Initio method, basis set incompleteness, and approximations inherent in the chosen density functional. This noise propagates into the MLIP model, limiting its achievable accuracy and potentially hindering its ability to generalize to configurations not present in the training set. Consequently, careful consideration of data quality, including error analysis and potential noise reduction techniques, is crucial for the successful application of MLIPs.
MACE (Machine learning for Atomic Configuartions and Environments) represents a class of machine learning interatomic potential (MLIP) frameworks utilizing equivariant neural networks. These networks are specifically designed to respect the rotational and translational symmetries inherent in atomic systems, allowing models to generalize effectively to configurations not explicitly present in the training data. By enforcing these symmetries directly within the network architecture, MACE-based MLIPs reduce the need for extensive data augmentation and achieve higher accuracy, particularly when extrapolating to new chemical environments or atomic arrangements. The equivariant property ensures that predictions remain physically meaningful regardless of the coordinate system used, a critical requirement for reliable molecular dynamics simulations.
Mitigating Noise: From Manual Intervention to Dynamic Approaches
Manual filtering techniques for noise reduction involve human inspection and selective removal of data points identified as erroneous or anomalous. While effective for small datasets, this process becomes increasingly impractical and costly as data volume grows due to the time required for individual assessment. The labor-intensive nature of manual filtering also introduces a scalability bottleneck, preventing its application to the large datasets common in modern machine learning and simulation contexts. Furthermore, the subjective element of human judgment can introduce inconsistencies and bias into the filtering process, potentially impacting the accuracy and reliability of subsequent analyses.
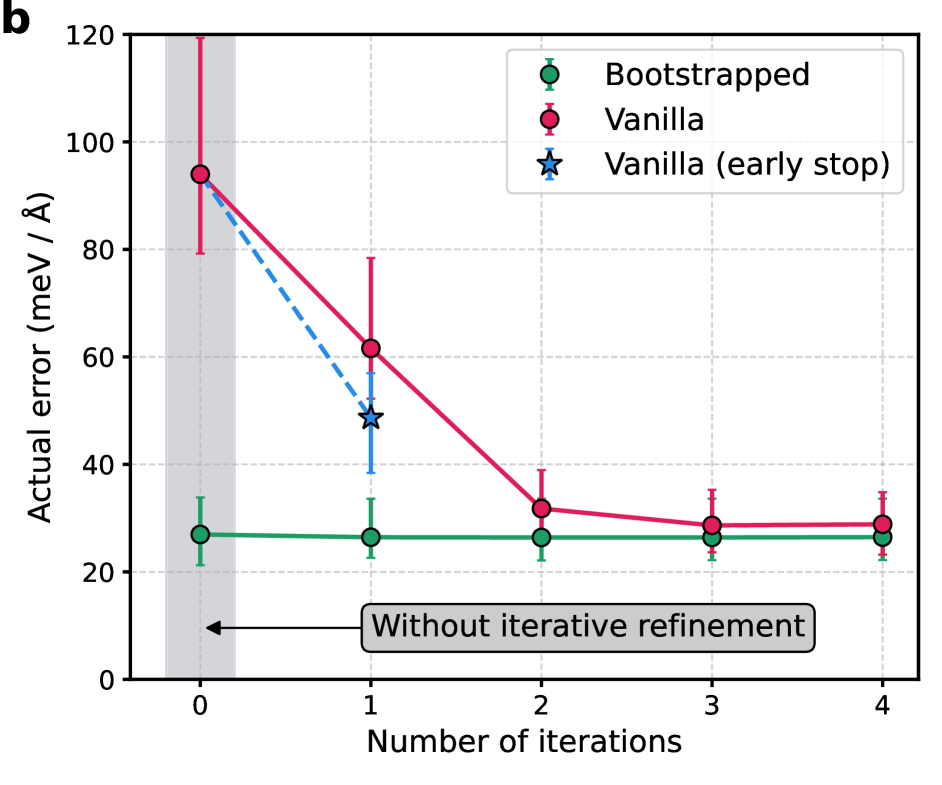
Iterative Refinement addresses the limitations of manual noise reduction by automating the process of data cleaning; however, this automation comes at a cost. The technique involves repeated cycles of model training followed by data filtering based on the model’s output. Each iteration necessitates a complete pass through the training data, increasing computational demands proportionally to the number of refinement cycles. While eliminating the labor of manual intervention, this repeated training and filtering introduces significant overhead, particularly when applied to large datasets or complex models, making it less efficient than single-pass dynamic approaches.
On-the-fly outlier detection, when integrated with exponential moving average (EMA) techniques, provides a dynamic noise mitigation strategy during model training. This approach avoids the need for pre-filtering or iterative refinement by continuously assessing data point likelihood during each training iteration. Suspect data points, identified as outliers based on pre-defined statistical thresholds or deviation from the EMA, are assigned reduced weights in the loss function. This down-weighting minimizes the influence of noisy data on model parameters without requiring separate filtering stages, resulting in a more computationally efficient training process and improved model accuracy.
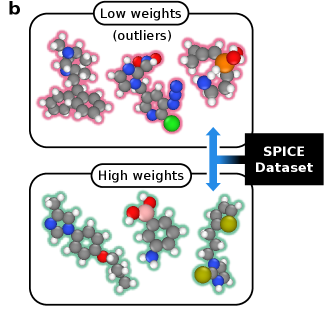
Dynamic Bootstrapping improves noise mitigation by modulating the contribution of each data point to the overall loss function based on its estimated likelihood. This adaptive weighting scheme prioritizes high-likelihood data while down-weighting potentially noisy or erroneous points during model training. Evaluations on the SPICE 2.0 dataset demonstrate a quantifiable benefit, with Dynamic Bootstrapping achieving a 3x reduction in energy errors compared to a baseline model without this adaptive weighting. This performance gain indicates that selectively focusing on more reliable data improves model accuracy and robustness in noisy environments.
Validation and Broad Applicability: A Robust Framework for Scientific Discovery
Rigorous validation is central to establishing the reliability of machine learning interatomic potentials (MLIPs); therefore, models incorporating noise mitigation techniques underwent testing against established benchmark datasets, notably the revMD17 Dataset and the SPICE 2.0 Dataset. Performance was quantitatively assessed using the root mean squared error (RMSE), a metric that effectively captures the average deviation between predicted and reference energies. This stringent evaluation process confirms the models’ ability to generalize beyond the training data, demonstrating their potential for accurate and dependable simulations in diverse materials science applications and paving the way for broader adoption within the scientific community.
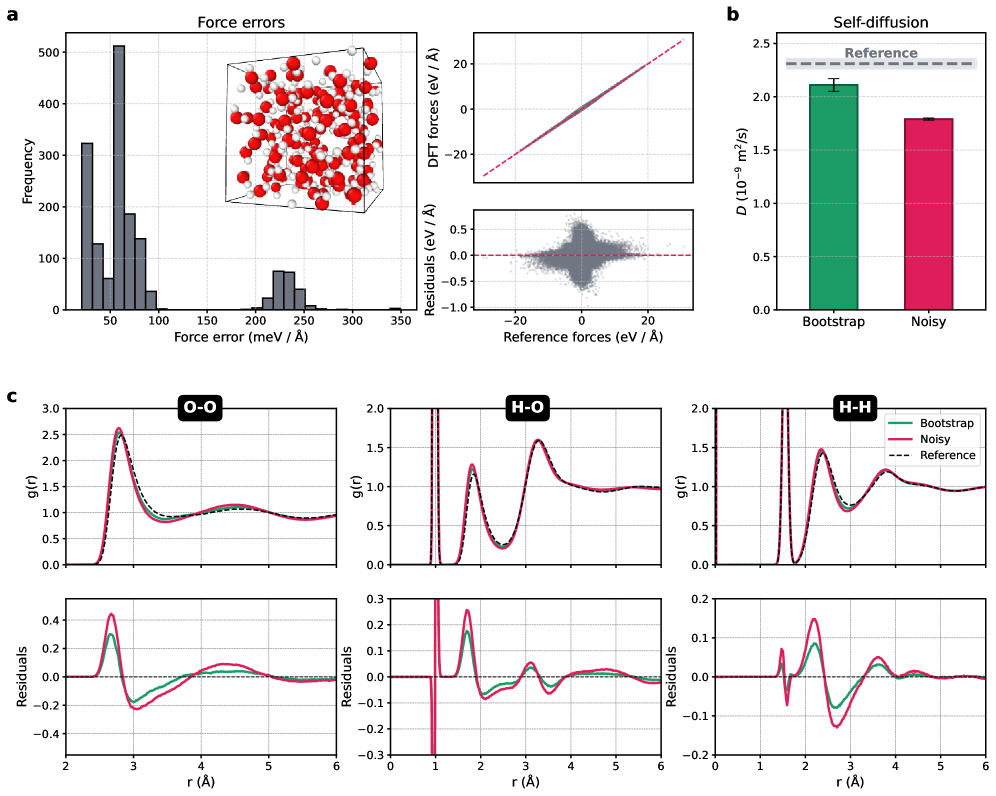
Rigorous validation of these machine learning interatomic potentials (MLIPs) reveals a remarkable level of accuracy, achieving a root-mean-square error (RMSE) of just 25-30 meV/Å when tested against the demanding revMD17 dataset. This performance signifies a substantial advancement in both predictive power and generalization ability, moving beyond simple memorization of training data to capture underlying physical principles. Consequently, these robust MLIPs facilitate dependable simulations of complex systems; for instance, water simulations utilizing this methodology yield a diffusion coefficient of 2.11 \times 10^{-9} m^2/s, differing by only 0.20 \times 10^{-9} m^2/s from established reference values, highlighting their potential to unlock new insights in materials science and chemistry.
Molecular dynamics simulations utilizing the developed machine learning interatomic potential accurately reproduce the diffusive behavior of water. Specifically, the calculated diffusion coefficient reached a value of 2.11 \times 10^{-9} \text{ m}^2/\text{s}, exhibiting a remarkably small deviation of only 0.20 \times 10^{-9} \text{ m}^2/\text{s} from established reference values. This level of agreement underscores the potential of this approach to reliably model complex aqueous systems, offering a crucial tool for investigations ranging from biomolecular processes to materials science applications where water plays a significant role.
The development of these noise-mitigation techniques transcends the limitations of individual datasets, establishing a foundational model with broad applicability across materials science and chemistry. Rather than requiring retraining for each new system, the underlying principles allow for transferrable learning, significantly reducing computational cost and accelerating discovery. This approach enables accurate predictions for materials and chemical processes beyond those used in initial training, fostering a paradigm shift towards more generalized and robust machine learning models. Consequently, researchers can leverage a single, well-trained model to investigate diverse phenomena, from predicting material properties to simulating complex chemical reactions, ultimately streamlining workflows and unlocking new avenues for scientific exploration.
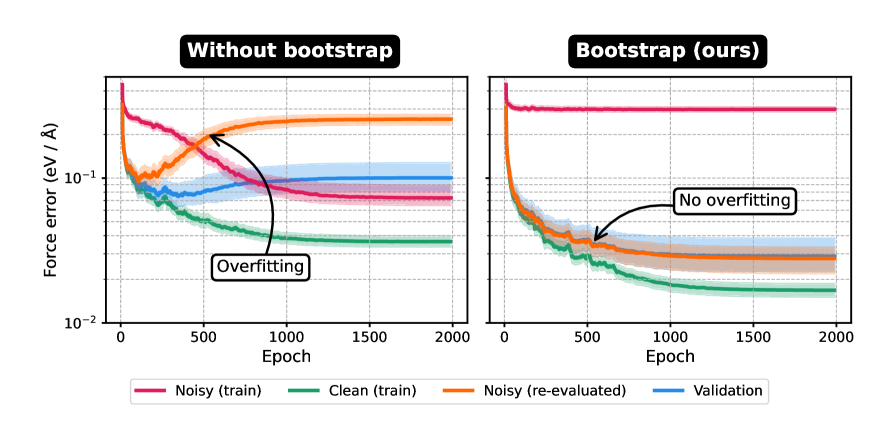
The pursuit of robust machine learning interatomic potentials, as detailed in this work, necessitates a rigorous filtering of aberrant data. The method presented actively addresses the inherent noise within training datasets, preventing its propagation through subsequent iterations. This echoes David Hume’s assertion: “A wise man proportions his belief to the evidence.” The dynamic bootstrapping approach mirrors this principle; it doesn’t blindly accept all data as truth, but rather assesses each point’s contribution to the overall model accuracy, discarding those that introduce instability. By focusing on verifiable evidence – in this case, consistent contributions to the potential energy surface – the training process becomes demonstrably more efficient and produces more reliable foundation models.
What’s Next?
The pursuit of robust machine learning interatomic potentials, as demonstrated by this work, continually highlights a fundamental tension: the reliance on data, and the inherent imperfection of that data. While dynamic bootstrapping offers a compelling defense against outlier influence, it merely addresses a symptom, not the disease. The true challenge lies not in filtering noise, but in constructing potentials inherently insensitive to it – a mathematical elegance currently elusive. Future work must explore methods that move beyond empirical resilience toward provable stability, perhaps leveraging techniques from robust optimization or differential geometry to constrain the potential energy surface.
The rise of foundation models in this domain amplifies the need for such rigor. Training on ever-larger datasets does not automatically confer correctness; it merely magnifies the impact of errors. The current paradigm, where model performance is assessed solely through empirical validation, remains fundamentally unsatisfying. A more principled approach would involve establishing formal guarantees on the potential’s accuracy and stability, independent of the training data itself.
In the chaos of data, only mathematical discipline endures. While practical improvements in outlier detection will undoubtedly continue, the ultimate goal must be a shift in focus: from chasing accuracy in the face of noise, to constructing systems where accuracy is defined by its immunity to it.
Original article: https://arxiv.org/pdf/2602.08849.pdf
Contact the author: https://www.linkedin.com/in/avetisyan/
See also:
- Gold Rate Forecast
- 22 Films Where the White Protagonist Is Canonically the Sidekick to a Black Lead
- Games That Faced Bans in Countries Over Political Themes
- Persona 5: The Phantom X Relativity’s Labyrinth – All coin locations and puzzle solutions
- How to Unlock Stellar Blade’s Secret Dev Room & Ocean String Outfit
- Celebs Who Narrowly Escaped The 9/11 Attacks
- Silver Rate Forecast
- ‘Super Mario Galaxy’ Trailer Launches: Chris Pratt, Jack Black, Anya Taylor-Joy, Charlie Day Return for 2026 Sequel
- 25 “Woke” Films That Used Black Trauma to Humanize White Leads
- 20 Films Where Black Directors Subverted Hollywood’s White Savior Tropes
2026-02-10 23:16